seed-pos
$ lexicmap utils seed-pos -h
Extract and plot seed positions via reference name(s)
Attention:
0. This command requires the index to be created with the flag --save-seed-pos in lexicmap index.
1. Seed/K-mer positions (column pos) are 1-based.
For reference genomes with multiple sequences, the sequences were
concatenated to a single sequence with intervals of N's.
So values of column pos_gnm and pos_seq might be different.
The positions can be used to extract subsequence with 'lexicmap utils subseq'.
2. All degenerate bases in reference genomes were converted to the lexicographic first bases.
E.g., N was converted to A. Therefore, consecutive A's in output might be N's in the genomes.
Extra columns:
Using -v/--verbose will output more columns:
len_aaa, length of consecutive A's.
seq, sequence between the previous and current seed.
Figures:
Using -O/--plot-dir will write plots into given directory:
- Histograms of seed distances.
- Histograms of numbers of seeds in sliding windows.
Usage:
lexicmap utils seed-pos [flags]
Flags:
-a, --all-refs ► Output for all reference genomes. This would take a long time for an
index with a lot of genomes.
-b, --bins int ► Number of bins in histograms. (default 100)
--color-index int ► Color index (1-7). (default 1)
--force ► Overwrite existing output directory.
--height float ► Histogram height (unit: inch). (default 4)
-h, --help help for seed-pos
-d, --index string ► Index directory created by "lexicmap index".
--max-open-files int ► Maximum opened files, used for extracting sequences. (default 512)
-D, --min-dist int ► Only output records with seed distance >= this value.
-o, --out-file string ► Out file, supports and recommends a ".gz" suffix ("-" for stdout).
(default "-")
-O, --plot-dir string ► Output directory for 1) histograms of seed distances, 2) histograms of
numbers of seeds in sliding windows.
--plot-ext string ► Histogram plot file extention. (default ".png")
-n, --ref-name strings ► Reference name(s).
-s, --slid-step int ► The step size of sliding windows for counting the number of seeds
(default 200)
-w, --slid-window int ► The window size of sliding windows for counting the number of seeds
(default 500)
-v, --verbose ► Show more columns including position of the previous seed and sequence
between the two seeds. Warning: it's slow to extract the sequences,
recommend set -D 1000 or higher values to filter results
--width float ► Histogram width (unit: inch). (default 6)
Global Flags:
-X, --infile-list string ► File of input file list (one file per line). If given, they are
appended to files from CLI arguments.
--log string ► Log file.
--quiet ► Do not print any verbose information. But you can write them to a file
with --log.
-j, --threads int ► Number of CPU cores to use. By default, it uses all available cores.
(default 16)
-
Adding the flag
--save-seed-posin index building.$ lexicmap index -I refs/ -O demo.lmi --save-seed-pos --force -
Listing seed position of one genome.
$ lexicmap utils seed-pos -d demo.lmi/ -n GCF_000017205.1 -o seed_distance.tsv $ head -n 10 seed_distance.tsv | csvtk pretty -t ref seqid pos_gnm pos_seq strand distance --------------- ----------- ------- ------- ------ -------- GCF_000017205.1 NC_009656.1 90 90 - 89 GCF_000017205.1 NC_009656.1 122 122 - 32 GCF_000017205.1 NC_009656.1 160 160 - 38 GCF_000017205.1 NC_009656.1 209 209 - 49 GCF_000017205.1 NC_009656.1 259 259 - 50 GCF_000017205.1 NC_009656.1 309 309 + 50 GCF_000017205.1 NC_009656.1 357 357 + 48 GCF_000017205.1 NC_009656.1 360 360 + 3 GCF_000017205.1 NC_009656.1 387 387 - 27Check the biggest seed distances.
$ csvtk freq -t -f distance seed_distance.tsv \ | csvtk sort -t -k distance:nr \ | head -n 10 \ | csvtk pretty -t distance frequency -------- --------- 126 1 99 32 98 36 97 40 96 36 95 40 94 37 93 48 92 62Or only list records with seed distances longer than a threshold.
$ lexicmap utils seed-pos -d demo.lmi/ -n GCF_000017205.1 -D 100 \ | csvtk pretty -t | head -n 5 ref seqid pos_gnm pos_seq strand distance --------------- ----------- ------- ------- ------ -------- GCF_000017205.1 NC_009656.1 168652 168652 + 126Plot histogram of distances between seeds and histogram of number of seeds in sliding windows.
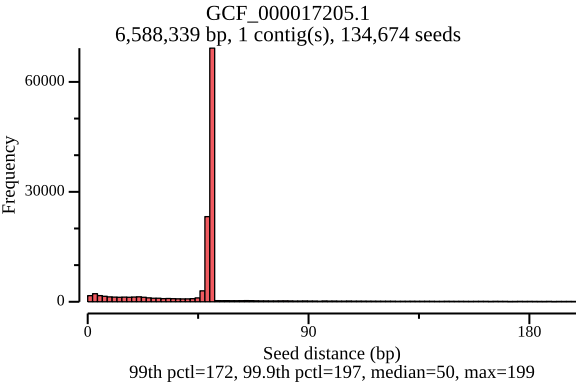
$ lexicmap utils seed-pos -d demo.lmi/ -n GCF_000017205.1 -o seed_distance.tsv --plot-dir seed_distance -w 250In the plot below, there’s a peak at 50 bp, because LexicMap fills sketching deserts with extra k-mers (seeds) of which their distance is 50 bp by default.
-
More columns including sequences between two seeds.
$ lexicmap utils seed-pos -d demo.lmi/ -n GCF_000017205.1 -v \ | head -n4 | csvtk pretty -t -W 40 --clip ref seqid pos_gnm pos_seq strand distance len_aaa seq --------------- ----------- ------- ------- ------ -------- ------- ---------------------------------------- GCF_000017205.1 NC_009656.1 90 90 - 89 9 TTAAAGAGACCGGCGATTCTAGTGAAATCGAACGGGC... GCF_000017205.1 NC_009656.1 122 122 - 32 3 TTTCTTTTAAAGGATAGAAGCGGTTATTGCTC GCF_000017205.1 NC_009656.1 160 160 - 38 3 TTGGTTGGACCGGTTTCTGTGTATAACTCATTGAAAGCOr only list records with seed distance longer than a threshold.
$ lexicmap utils seed-pos -d demo.lmi/ -n GCF_000017205.1 -v -D 100 \ | csvtk pretty -t -W 40 ref seqid pos_gnm pos_seq strand distance len_aaa seq --------------- ----------- ------- ------- ------ -------- ------- ---------------------------------------- GCF_000017205.1 NC_009656.1 168652 168652 + 126 0 GGCGGCGTCGGCGGCGCCACGCTCGCTGGCTGTGGCTGTG GCTGTGGCTGTGGCTGTGGCTGTGGCTGTGGCTGTGGCTG TGGCTGTGGCTGTGGCTGTGGCTGTGGCGGCTGCTGGGTG ATCCCGIt appears to be a highly repetitive region, specifically a tandem repeat with the unit sequence
CTGTGG:$ lexicmap utils seed-pos -d demo.lmi/ -n GCF_000017205.1 -v -D 100 \ | csvtk cut -t -f seqid,seq \ | csvtk del-header -t \ | seqkit tab2fx \ | seqkit locate --only-positive-strand --non-greedy --pattern CTGTGG \ | csvtk pretty -t seqID patternName pattern strand start end matched ----------- ----------- ------- ------ ----- --- ------- NC_009656.1 CTGTGG CTGTGG + 30 35 CTGTGG NC_009656.1 CTGTGG CTGTGG + 36 41 CTGTGG NC_009656.1 CTGTGG CTGTGG + 42 47 CTGTGG NC_009656.1 CTGTGG CTGTGG + 48 53 CTGTGG NC_009656.1 CTGTGG CTGTGG + 54 59 CTGTGG NC_009656.1 CTGTGG CTGTGG + 60 65 CTGTGG NC_009656.1 CTGTGG CTGTGG + 66 71 CTGTGG NC_009656.1 CTGTGG CTGTGG + 72 77 CTGTGG NC_009656.1 CTGTGG CTGTGG + 78 83 CTGTGG NC_009656.1 CTGTGG CTGTGG + 84 89 CTGTGG NC_009656.1 CTGTGG CTGTGG + 90 95 CTGTGG NC_009656.1 CTGTGG CTGTGG + 96 101 CTGTGG NC_009656.1 CTGTGG CTGTGG + 102 107 CTGTGGA similar case in another genome.
$ lexicmap utils seed-pos -d demo.lmi/ -n GCF_003697165.2 -v -D 100 \ | csvtk pretty -t -W 40 ref seqid pos_gnm pos_seq strand distance len_aaa seq --------------- ------------- ------- ------- ------ -------- ------- ---------------------------------------- GCF_003697165.2 NZ_CP033092.2 1563265 1563265 - 202 29 TAAGACTCAAGACTCAAGACTCAAGACTCAAGACTCAAGA CTCAAGACTCAAGACTCAAGACTCAAGACTCAAGACTCAA GACTCAAGACTCAAGACTCAAGACTCAAGACTCAAGACTC AAGACTCAAGACTCAAGACTCAAGACTCAAGACTCAAGAC TCAAGACTCAAGACTCAAGACTCAAGACTCAAGACTCAAG AC -
Listing seed position of all genomes.
$ lexicmap utils seed-pos -d demo.lmi/ --all-refs -o seed-pos.tsv.gzShow the number of seed positions in each genome.
$ csvtk freq -t -f ref -nr seed-pos.tsv.gz | csvtk pretty -t ref frequency --------------- --------- GCF_000017205.1 143165 GCF_000742135.1 120758 GCF_003697165.2 110132 GCF_000006945.2 108387 GCF_002950215.1 108272 GCF_002949675.1 101098 GCF_009759685.1 88632 GCF_000392875.1 65403 GCF_001027105.1 64176 GCF_001544255.1 57167 GCF_006742205.1 57086 GCF_001096185.1 49482 GCF_900638025.1 48959 GCF_001457655.1 45771 GCF_000148585.2 44752Plot the histograms of distances between seeds for all genomes.
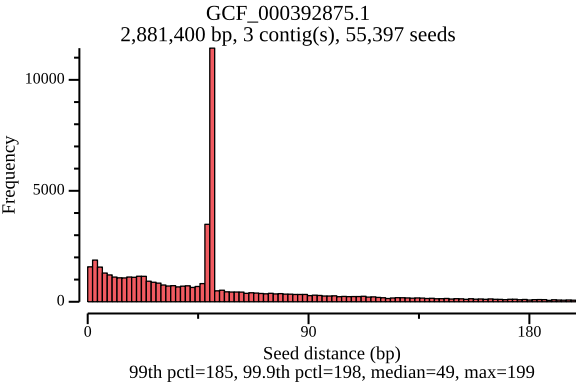
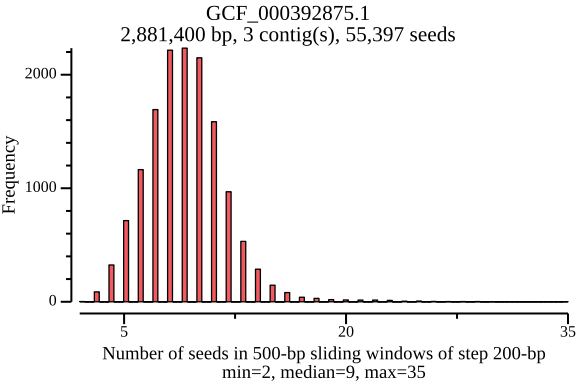
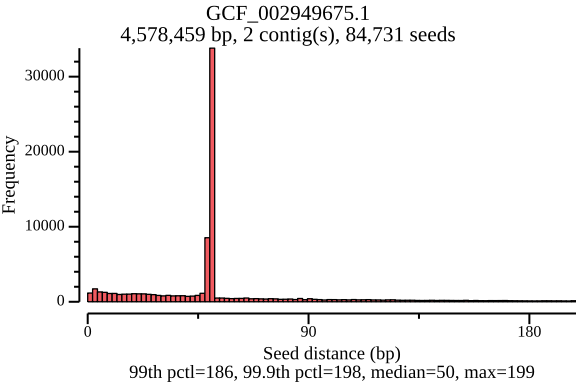
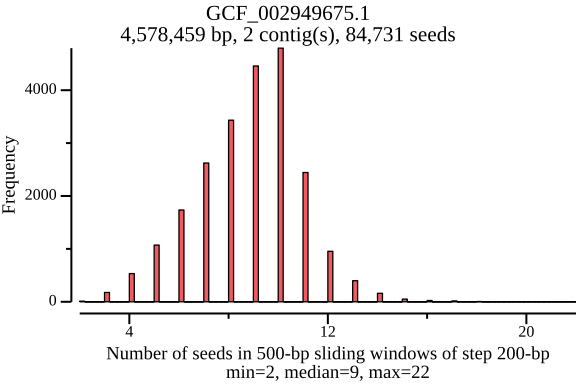
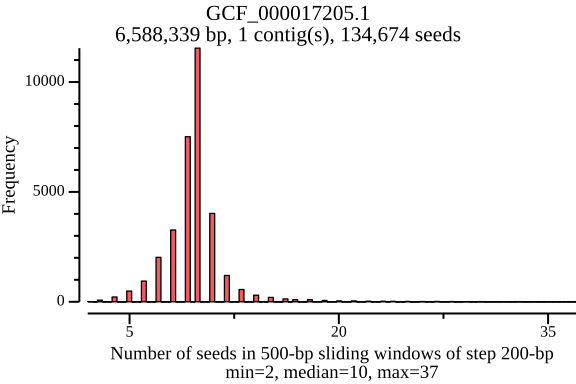
$ lexicmap utils seed-pos -d demo.lmi/ --all-refs -o seed-pos.tsv.gz \ --plot-dir seed_distance --force 09:56:34.059 [INFO] creating genome reader pools, each batch with 1 readers... processed files: 15 / 15 [======================================] ETA: 0s. done 09:56:34.656 [INFO] seed positions of 15 genomes(s) saved to seed-pos.tsv.gz 09:56:34.656 [INFO] histograms of 15 genomes(s) saved to seed_distance 09:56:34.656 [INFO] 09:56:34.656 [INFO] elapsed time: 598.080462ms 09:56:34.656 [INFO] $ ls seed_distance/ GCF_000006945.2.png GCF_000742135.1.png GCF_001544255.1.png GCF_006742205.1.png GCF_000006945.2.seed_number.png GCF_000742135.1.seed_number.png GCF_001544255.1.seed_number.png GCF_006742205.1.seed_number.png GCF_000017205.1.png GCF_001027105.1.png GCF_002949675.1.png GCF_009759685.1.png GCF_000017205.1.seed_number.png GCF_001027105.1.seed_number.png GCF_002949675.1.seed_number.png GCF_009759685.1.seed_number.png GCF_000148585.2.png GCF_001096185.1.png GCF_002950215.1.png GCF_900638025.1.png GCF_000148585.2.seed_number.png GCF_001096185.1.seed_number.png GCF_002950215.1.seed_number.png GCF_900638025.1.seed_number.png GCF_000392875.1.png GCF_001457655.1.png GCF_003697165.2.png GCF_000392875.1.seed_number.png GCF_001457655.1.seed_number.png GCF_003697165.2.seed_number.pngIn the plots below, there’s a peak at 50 bp, because LexicMap fills sketching deserts with extra k-mers (seeds) of which their distance is 50 bp by default. And they show that the seed number, seed distance and seed density are related to genome sizes.
-
GCF_000392875.1 (genome size: 2.9 Mb)
-
GCF_002949675.1 (genome size: 4.6 Mb)
-
GCF_000017205.1 (genome size: 6.6 Mb)
-
The output (TSV format) is formatted with csvtk pretty. SeqKit is used to locating subsequences from fasta files. lexicmap utils subseq can also be used to extract subsequences from the index.